
Acting as enzymes, structural elements, and molecular motors, proteins represent the molecular drive of biological systems, the elucidation of which relies on the explication of conformational transitions. In a recent study by Linköping University, Sweden, researchers got into the issue of protein conformational sampling, stating that there are limitations to current methods and that AFsample2 is one of the few and far methods doing a good job at generating models as proteins are diverse.
Challenge of Multiple Conformations
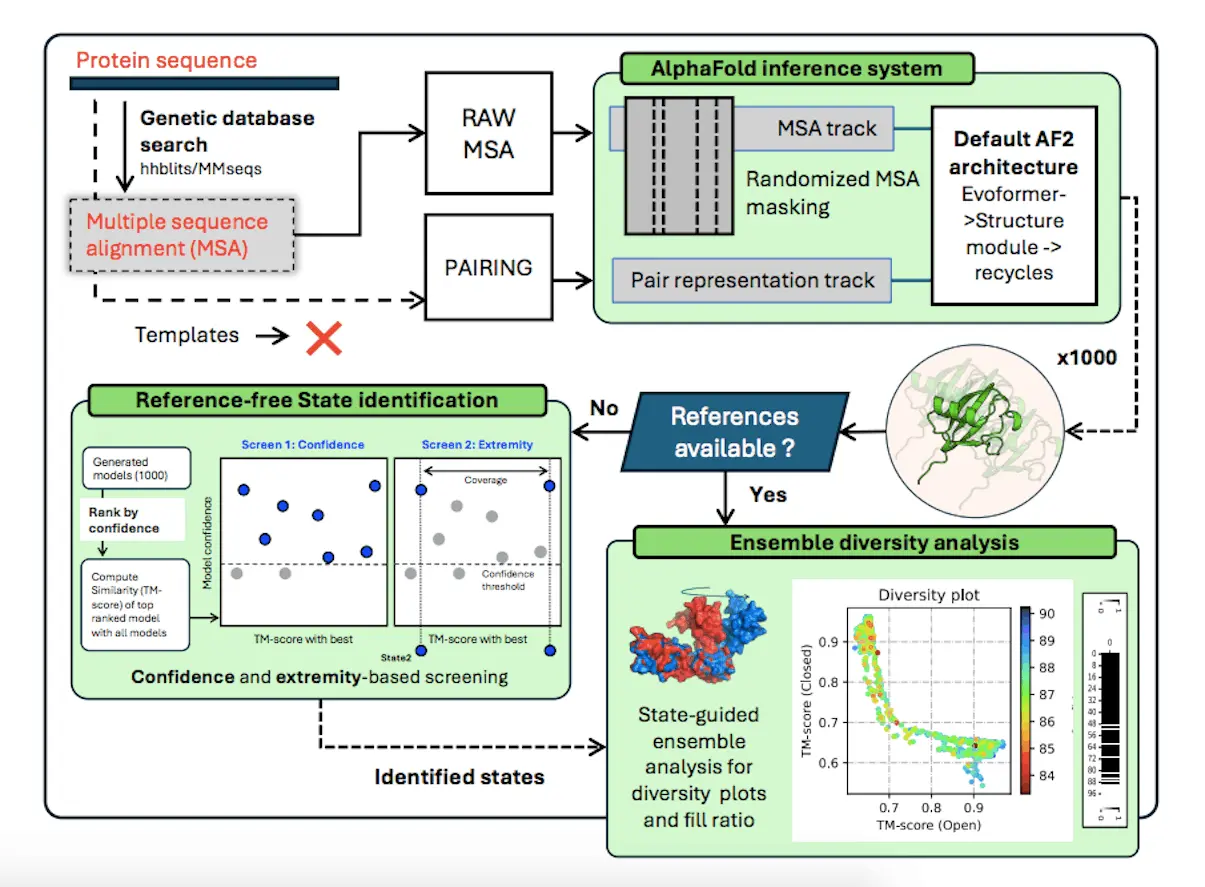
One of the most difficult problems in protein structure prediction is predicting multiple conformations after the optimization of the standard folding algorithms trained for single-state models. The difficulties stressed in the article raise whether current approaches to estimate uncertainties based on model confidence can correctly model the alternative state in many cases because of the restrictions inherent within the inference system. Researchers propose a solution named AFsample2, which incorporates key modifications such as randomized MSA column masking to increase conformational space sampling and deliver better estimations of the distinct conformations. By alleviating co-evolutionary constraints, AFsample2 provides a way to improve the fold-switch proteins, deceiving conformational variability, which could further help in the development of the protein structure prediction field.

Source: https://github.com/iamysk/AFsample2/
Adaptability and Performance
Apart from single-state proteins, AFsample2 suggests possible compatibility with the multiprotein system, which opens up prospects for the further use of the complex in structural biology. Based on such qualities, the tool is likely beneficial for enhancing conformational samples and can be potentially effective for improving future protein structure prediction in biological systems.
Performance Analysis
AFsample2 performs significantly better than other traditional methods such as AFvanilla and AFdropout, clearly displaying higher model efficiency and the capacity to continually produce good quality protein models on all types of proteins quickly. With all Z-scores being positive values, it is clear that the movement that has been sampled corresponds well with the expected result, making the tool rather effective in predicting protein structures.
Comparing AFsample2 to AFvanilla, AFdropout, and 217 AFcluster
Open-closed conformations (OC23) dataset was used for comparison with AFsample2 and standard AF2 (AFvanilla), AF2 with dropout (AFdropout), and AFcluster, the models of which were generated from 1000 iterations for each protein. AFvanilla and AFdropout were similar in terms of RasMdist with narrow distributions that were near high TM-score for the closed state, while AFsample2 had a relatively wider distribution for both opened and closed states. However, there were obstacles in building the best models for both states within AFcluster, while the advanced conformational sampling and diversity of AFsample2 demonstrated its advantage.
Modifying AlphaFold2 for Ensemble Predictions
AlphaFold2 allows for the concept of ensemble prediction by making alterations in the Gaussian inference procedure, including random sanitization of the multiple sequence alignment column and other new methods to assess the conformations independently of having a reference experimental structure. Altogether, it demonstrates that by adding noise to the MSA feature profile and improving the identification of other states, AlphaFold2 can provide accurate conformational states for various proteins. These improvements allow AlphaFold2 to enlarge the conformational space of the predictions and give researchers more opportunities to outline the structural dynamics of proteins comprehensively.
Application and Implication
It is clear that integrating modifications for AlphaFold2 towards ensemble prediction holds myriad potential applications and implications. By improving conformational space exploration of the structure, much more information about protein dynamics, functions, and interactions could be obtained. They broadly impact drug design, protein design, and functions of protein at molecular levels. Thus, employing accurate multiple conformational states can help enrich new prospects of investigating multifaceted biological processes and creating effective drugs. In conclusion, the proposed modification in AlphaFold2 allows for combining deep learning and simple physics to drive the protein folding process, so it can be used in various fields for solving structural biology problems.
Future Direction
Future work for the AFsample2 is to continue more research on the ensemble predicted methods aiming at enhanced preciseness and time consumption. Future improvements may, therefore, target the identification of new approaches to assess the accuracy of predicted conformational states, as well as the enhancement of the tool’s transferability to additional protein systems. Furthermore, investigating the simultaneous application of other structural biology methods like cryo-EM and NMR spectroscopy to further complement the assessment of protein structure and dynamics could also improve the understanding of structures within this dataset. It means that further development of the ensembles and the use of AlphaFold2 are set to change the direction of structural biology and stimulate diverse inventions in drug development and biotechnology.
Conclusion
AFsample2 ensemble prediction represents a key development in several realms, resulting in improved protein structure prediction and conformational sampling. Because of the possibility of predicting multiple states simultaneously and owing to the new approaches adopted by AlphaFold2 to span conformational space better, the stage has been set for making the next qualitative leap towards dissecting self-assembly processes and understanding protein function. This modified tool has potential uses where its strategies have already been applied, namely in drug discovery, protein engineering, and structural biology, to assist in answering biological questions and moving scientific knowledge forward. Further, even the ongoing research in this area offers tremendous prospects for the future of structural biology and many discoveries in the field of life sciences.
Article Source: Reference Paper | Data Availability: GitHub
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anshika is a consulting scientific writing intern at CBIRT with a strong passion for drug discovery and design. Currently pursuing a BTech in Biotechnology, she endeavors to unite her proficiency in technology with her biological aspirations. Anshika is deeply interested in structural bioinformatics and computational biology. She is committed to simplifying complex scientific concepts, ensuring they are understandable to a wide range of audiences through her writing.











{kind=link}
[…] Predicting Multiple Conformations and Ensembles with AFsample2: An AlphaFold2 Extension […]