
Although molecular dynamics (MD) simulations provide accurate representations of the motion of molecular systems, molecular biology and materials science are challenged by the computational demands of these simulations. Since deep learning techniques have proven successful in many domains, it is unclear if they can be used to improve the effectiveness of MD simulations. Researchers from the University of Cambridge created Molecular Dynamics Language Models (MDLMs) to explore this possibility and make it possible to generate MD trajectories. Using kernel density estimations from large MD datasets, the current MDLM implementation maintains structural correctness while training on a brief classical MD trajectory of a tiny protein. By identifying conformational states that were undersampled in the training data, this method makes it possible to determine the protein’s free energy landscape. It offers preliminary proof of the effective application of molecular dynamics.
Introduction
Utilizing kernel density estimations from large MD datasets, the current MDLM implementation maintains structural correctness while training on a brief classical MD trajectory of a tiny protein. By identifying conformational states that were undersampled in the training data, this method makes it possible to determine the protein’s free energy landscape. It offers preliminary proof of the effective application of molecular dynamics.
Molecular dynamics (MD) simulations are the gold standard for studying protein dynamics utilizing physical rules of motion. By integrating motion equations, these simulations explore the conformational space of proteins in atomic-level detail. However, it frequently takes a lot of time and processing power to achieve convergence in MD simulations, especially when studying infrequent occurrences or high-energy states. Rare structural changes and long-timescale events are difficult to examine due to this computational load. The difficulty is in striking a balance between conflicting needs like conformational diversity accuracy, computational efficiency, and respect for physical principles by fusing the effectiveness of machine learning with the physical correctness of MD simulations.
Since Language Models (LMs) are effective tools for pattern identification and generation, they present opportunities to overcome the difficulties associated with producing MD trajectories that embody the fundamental laws of motion. In general, one can inquire whether LMs can capture the grammar or the physical laws of protein movements by modeling proteins as words and MD trajectories as sentences.
Understanding Molecular Dynamics Language Models
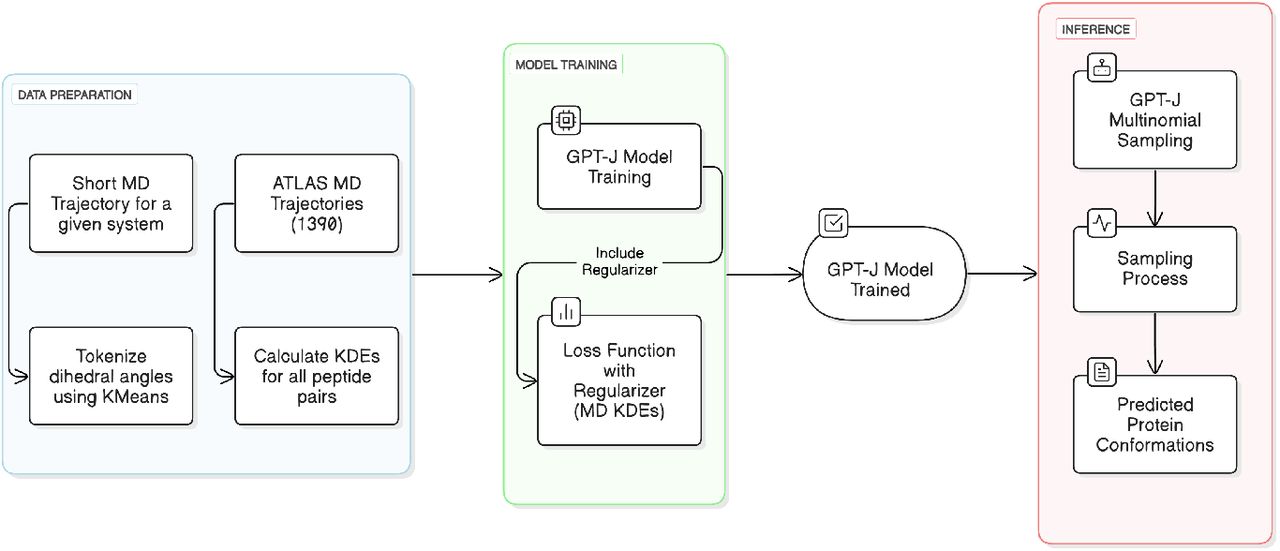
The researchers here describe Molecular Dynamics Language Models (MDLMs), a framework that connects the pattern recognition capabilities of LMs with the physical accuracy of MD simulations, to explore such prospects. Researchers use a combination of general physical concepts and system-specific learning. MDLMs are efficient and accurate because they incorporate physical insights from large MD datasets while training on a small fraction of the generally required MD simulation time for a particular system. To execute this physical guidance, 1390 MD trajectories from the ATLAS database are used to create kernel density estimations (KDEs), which represent permitted conformational regions.
Application of Molecular Dynamics Language Models
The application of MDLMs, which researchers described, shows that it can effectively investigate a protein’s conformational space while preserving physical precision. It provides notable improvements in conformational sampling using only 5% of MD trajectory data, with Jaccard indices improving in difficult instances like the PE residues pair. Important thermodynamic characteristics, such as the proper folded-unfolded energy differential (1.5 kcal/mol) and transition barriers (2.0 kcal/mol), are captured by the reconstructed free energy landscape and agree with the entire trajectory. With only moderate processing resources, the MDLM exhibits strong performance across various peptide combinations despite the existence of some difficult instances, especially for highly flexible sequences like GY. This work serves as a proof of concept for MDLMs and is centered on backbone conformations.
Language Models for Protein Structure Prediction and Design
Language models have enabled the prediction and design of protein structures, especially those based on the transformer architecture, such as the generative pre-trained transformer (GPT) series. These causal models make sure that every predicted structural element takes into account the complete previous structural context, which is consistent with the sequential pattern of conformational changes in proteins. Language models ‘ rotational positional embeddings offer an intense method for comprehending relative locations in sequences, which enables the model to process and produce conformational changes while preserving structural coherence. The beam search multinomial sampling technique is essential in language model inference to make a variety of physically realistic conformations and enable controlled exploration of the conformational space.
Future Direction
To improve accuracy and handle side-chain interactions more effectively, future developments will expand to include all-atom representation. Further performance improvement could be achieved by physical guidance and sample process optimization, especially for complex cases like highly flexible residues.
Conclusion
The use of deep learning techniques in structural biology has resulted in revolutionary methods for determining protein structures since it is now feasible to predict protein structures from amino acid sequences with an accuracy that is often comparable to high-resolution experimental techniques. Recent findings that support using deep learning techniques to describe the conformational space of proteins in terms of Boltzmann ensembles have proved encouraging. However, applying deep learning techniques to the prediction of protein dynamics is still an outstanding subject. The amino acid sequence of the protein being studied can be used to train large language models (LLMs) on an extensive range of MD trajectories to create protocols for executing LLM-based MD simulations as research advances.
Article Source: Reference Paper.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}