
A significant issue in genome research is the Nucleotide Transformer, which makes DNA sequence interpretation accurate only with models such as DNABERT-2. Although these models have made great strides in a number of DNA analysis tasks, issues with genetic language variety, model efficiency, and length extrapolation remain. The objective is to improve performance at costs that are scalable and to correctly interpret sequences of any length inside a single model framework. In response, Southeast University researchers present DeepGene, a model that uses Minigraph and Pan-genome representations to capture the wide range of genetic linguistic variety. DeepGene uses rotational position embedding to enhance length extrapolation in various genetic analysis applications. DeepGene scores the highest overall on the 28 tests in the Genome Understanding Evaluation, placing first in nine of them and second in five others. DeepGene performs better than other state-of-the-art models due to its small model size and increased processing efficiency for sequences of different lengths.
Introduction
In many ways, DNA is comparable to natural language in that it is a biological language. Human DNA sequence modeling as a natural language paradigm has a lot of potential right now. An abundance of large-scale models trained on genetic corpora has evolved with the introduction of Transformer-based models. Utilizing continuous embeddings, these models simulate DNA sequences and show promise for use in a range of genomic analytic tasks, such as predicting promoters, deciphering enhancer complexity, comprehending gene regulation, and projecting gene expression.
Genomic foundation models like DNABERT, DNABERT-2, and Nucleotide Transformer have demonstrated cutting-edge results in various genomics activities.
Using K-mer based sequence tokenization, DNABERT pre-trained on the human reference genome GRCh38.p13. This method worked incredibly well for applications like transcription factor binding site identification and promoter prediction. In a follow-up study, DNABERT-2 was converted to Byte Pair Encoding (BPE) for DNA segmentation, and the size of its training dataset was increased to include genomes from other species. Furthermore, DNABERT-2 set the standard for the Genome Understanding Evaluation (GUE) by outperforming other top foundation models. The Nucleotide Transformer was notable for utilizing large data volumes and model sizes, having been pre-trained on a blend of human pan-genome and multi-species genomes. In addition, a large number of different pre-trained models for modeling DNA sequences have been developed.
Challenges Faced by Foundational Models
- Reference Genome vs. Pan-Genome: Reference genomes served as the main training set for DNABERT and DNABERT-2. However, a single reference genome cannot fully reflect the scope of genetic variation within a species, as does the notion of the pan-genome. It includes all of the changes found in the DNA language, including single nucleotide polymorphisms (SNPs), insertions, deletions, and structural differences between different people or groups. A more thorough comprehension of species-specific genetic variation is made possible by this variety.
- Performance vs. Costs: Larger genomic models typically perform better, but the gains become less significant as the model gets bigger and more computer resources are needed. Achieving the ideal balance between cost and performance is essential for effectively training genomic models on large-scale DNA datasets.
- Short Sequence vs. Long Sequence: Accurate genomic sequence decoding necessitates comprehending copious contextual information dispersed over thousands of nucleotides. DNABERT and Nucleotide Transformer had maximum input sequence sizes of 512 bps and 1000 bps, respectively. With sliding attention, Gena-LM increased the length of the input sequence to between 4.5k and 36k base pairs, demonstrating better length extrapolation capabilities than earlier models. On the other hand, Gena-LM has trouble analyzing brief sequences well.
Understanding DeepGene
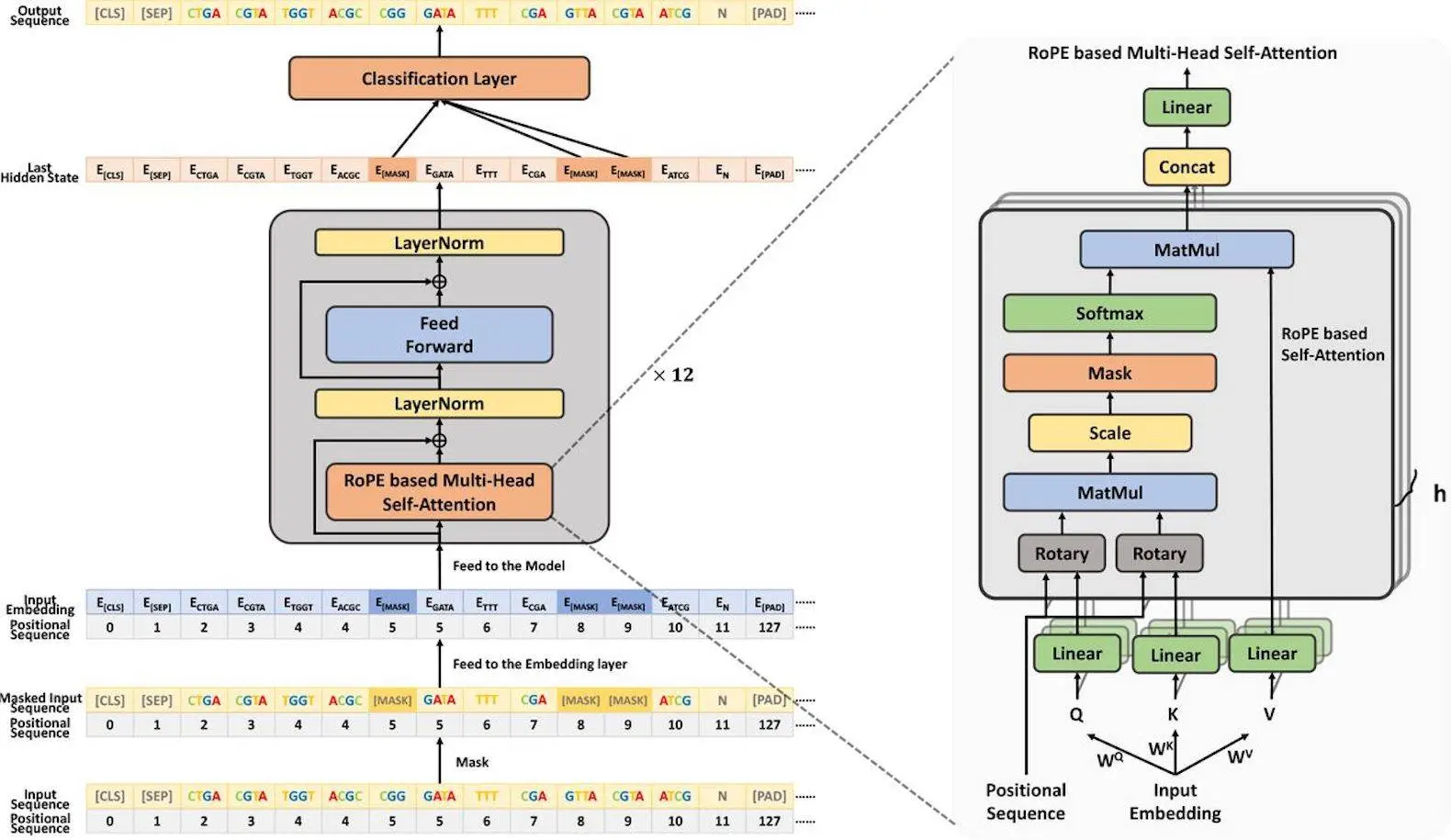
DeepGene is a Transformer-based architecture designed to segment DNA via BPE tokenization. The innovative contributions are as follows:
- A large dataset consisting of 47 ancestrally distinct human pan-genomes is used to train DeepGene. This dataset allows DeepGene to capture the extensive heterogeneity of genomic language to provide a more nuanced view of genetic variety.
- Additionally, the researchers use the Minigraph representation to effectively describe the complex graph structures inherent in pan-genomes.
- DeepGene uses rotary position embedding to precisely learn the relative positions of bases. This improves the model’s performance for length extrapolation, making it suitable for both short and large DNA sequences. Its remarkable ability to decipher and predict genomic sequences has been shown in experiments across a wide range of tasks and datasets, indicating its potential to provide important new insights into the intricate language of DNA.
About Pan-genome Dataset
The reference genomes used to train DNABERT and DNABERT-2 were unable to fully capture the range of genetic variants. The Nucleotide Transformer’s base model was learned using samples from thousands of different human genomes in addition to a reference genome. This strategy introduced a 98% redundancy rate among the samples, but it also greatly increased the dataset. The Nucleotide Transformer’s capacity to comprehend the variety of genetic language was limited by this high degree of redundancy.
In genomic investigations, the term “pan-genome” has come to refer to population diversity. It provides a deeper reservoir of genetic variety in contrast to conventional reference genomes. This increased variety makes it possible to comprehend more precisely and effectively the traits of genetic language. Pan-genome corrects the biases of reference genomes by keeping a representative set of various haplotypes and their alignments. This has resulted in the development of various pan-genomic graph models, such as variation graphs and de Bruijn graphs. Because graph topologies naturally provide advantages, they are now the most exceptional at managing big volumes of incoming data. A pan-genome graph can eliminate duplication. The multiple sequence alignment approach addresses the pan-genomic data issue and enables the model to concentrate more on the distinct components. De Bruijn graphs are made up of k-mers from all haplotypes; a k-mer is represented by each node, and there are k-1 overlapping bases between two connected nodes.
In contrast, variation graphs store each input haplotype using sequence graphs and path lists rather than having a fixed base length on each node. Researchers select the variation graphs for two reasons: (1) Due to k-mer tokenization, there is a great deal of overlap in De Bruijn graphs between nodes. (2) The variation graph can be further tokenized because of the nodes’ varying lengths.
The Human Pangenome Reference Consortium recently presented a draft of the human pan-genome. The Pan-genome Graph Builder (PGGB), Minigraph, and Minigraph-Cactus techniques were used to generate the pan-genome graph shown in this research, which was based on the sequencing findings of 47 ancestrally diverse people.
Limitations of the Work
Tokenization and edge quality in the pan-genomic graph of the Minigraph model need to be improved. The multi-species pan-genomic data might be more efficient, and the model’s BPE tokenization technique needs to be improved. It is unreasonable to rely exclusively on the model’s limited extrapolation capability for longer runs. The attention layer must be modified to include sparse attention, among other things, to increase the model’s capacity to handle longer sequences.
Future Work
Future research will concentrate on finding ways to enhance large-scale language models. For pre-training, the Nucleotide Transformer employs a pan-genome dataset containing more than a thousand individuals, necessitating linear time complexity for lengthy sequences. Combining genomes from several animals can improve the skill of the model. These methods seek to advance the use, creation, and enhancement of language models based on genetics.
Conclusion
The model’s superiority in length extrapolation is demonstrated by the 6mer’s somewhat better performance than the 3mer’s. This illustrates how well the model can represent longer sequences. The model, which is based on the Minigraph, uses processes including DAG building, BPE tokenization, and serialization to extract token and positional information from pan-genomic graph data. The model is trained with relative positional encoding in order to enhance its length extrapolation capability. On the GUE evaluation criteria, the model performs comparably even with fewer training data and a shorter training period. LPD studies demonstrate that the model performs better than DNABERT with absolute positional encoding in half of the challenges.
Article Source: Reference Paper | The datasets and source code of DeepGene are available on GitHub.
Important Note: bioRiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}