
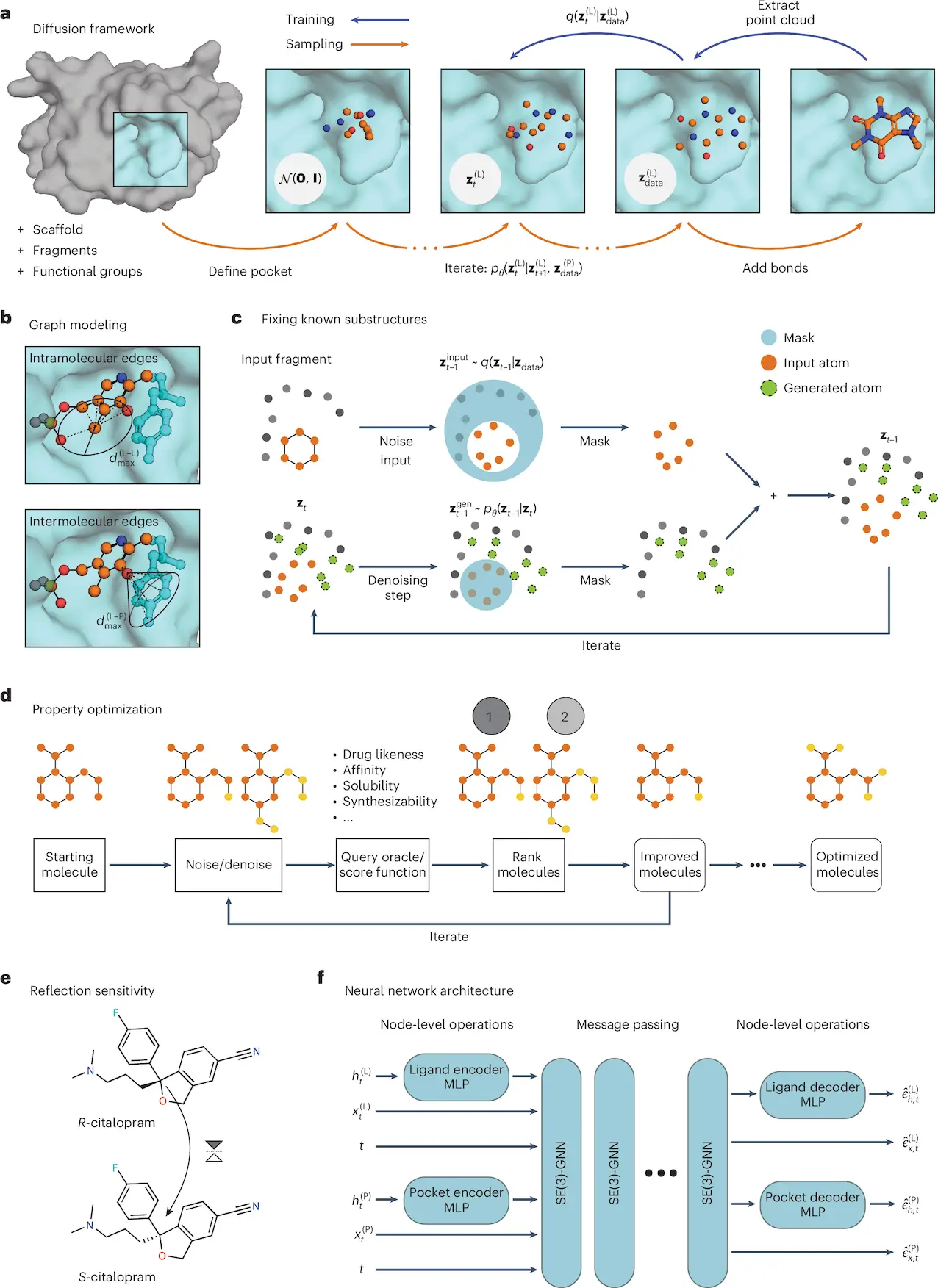
In the quest to create better and faster methods for drug discovery, a team of innovative researchers from various institutions has developed a groundbreaking technique that leverages equivariant diffusion models for structure-based drug design (SBDD). This study, published in Nature Computational Science by École Polytechnique Fédérale de Lausanne, Switzerland, addresses the growing need for computational tools to predict molecular structures with high accuracy, specificity, and diversity—a crucial step in developing effective therapeutics. Here researchers present DiffSBDD, a SE(3)-equivariant diffusion model that produces new ligands conditioned on protein pockets.
The Need for Advanced Computational Models in Drug Design
Discovering drugs takes a lot of effort and can be extremely expensive. The classical approaches ensure that considerable trial and error is required to identify molecules that could interact with the protein target of interest with the best affinity and the necessary pharmacological and chemical characteristics. New approaches like computational biology have enabled the change to this paradigm. Still, existing models have the challenge of achieving a proper balance between molecular diversity and structural accuracy, especially in novel protein targets.
This is where the research team’s new approach contributes. With equivariant diffusion models and generative design, they built DiffSBDD-cond, a model that learns to create active ligands for a protein appropriate to the target. Their research proposes an excellent approach for scaffold hopping, fragment merging, and repetitive evolution that is expected to improve the efficiency of drug discovery workflows.
What Is Structure-Based Drug Design?
At its core, structure-based drug design (SBDD) involves creating drug molecules by analyzing the three-dimensional structure of a protein target. All proteins are important biomolecules carrying different biological processes, whereas certain diseases are a consequence of a protein’s dysfunction. Drugs act on these proteins to change their activity so that the normal restoration function is resumed or adverse effects are eliminated.
The primary task here is to conceive of drug molecules that will attach to the protein’s active site with precision. For this, it is always a matter of compromise: the extent to which a molecule binds satisfactorily, its shape may fit into the protein, its chemistry, and its foldability as an effective drug. This situation has been improving with traditional SBDD, but the complexity of molecular interactions and chemical space remains a challenge.
Introducing DiffSBDD-cond
The study introduces DiffSBDD-cond, a diffusion model trained on a dataset of protein-ligand complexes. Diffusion models are a class of artificial intelligence techniques that are used primarily for inserting images, but because of their versatility, they have found applications in molecular design as well. What makes DiffSBDD-cond unique is that it possesses a property known as equivariance — the ability to remain constant even when delivered with a modification such as a rotation or translation. This particular property guarantees that molecular architectures are indeed constructed in a precise manner within the three-dimensional binding cavity of a protein.
The model is quite good at understanding the physical and chemical features surrounding the protein-ligand contact region. It presents a suitable strategy for designing molecules that are pharmacologically active while maintaining a certain degree of freedom in varying molecular designs. Now, let us focus on the particular challenges DiffSBDD-cond solves.
Molecular Inpainting: Enhancing Drug Design
The researchers demonstrated the model’s capabilities through five core molecular inpainting tasks:
- Scaffold Hopping
- Objective: Replace a drug’s backbone structure (scaffold) while retaining functional groups responsible for binding.
- Example: DiffSBDD-cond redesigned inhibitors for mitotic kinesin Eg5 (PDB: 2gm1), a protein involved in cell division.
- Scaffold Elaboration
- Objective: Expand an existing scaffold by adding new functional groups to improve drug efficacy.
- Example: A rationally designed inhibitor for the ENAH EVH1 protein (PDB: 6rcj) served as a test case.
- Fragment Merging
- Objective: Combine small fragments identified experimentally into a cohesive molecule.
- Example: Fragments binding to SARS-CoV-2 non-structural protein 3 (Nsp3) (PDB: 5rsw, 5rue) were merged, replicating prior research results.
- Fragment Growing
- Objective: Extend a central motif of a molecule by designing new molecular extensions.
- Example: Molecules targeting ENAH EVH1 (PDB: 5ndu) were grown to optimize interactions.
- Fragment Linking
- Objective: Design new fragments with connecting linkers to bridge isolated parts of a molecule.
- Example: The model created linkers for ENAH EVH1 inhibitors (PDB: 5ndu).
Each of these tasks showcases the model’s ability to maintain high chemical complementarity and binding accuracy while offering novel design possibilities.
Iterative Optimization for Better Molecules
To further refine the molecules, the researchers came up with an iterative optimization process. This method starts by perturbing a solvent to coat the structure of a known molecule, producing a diversity of candidates around its chemical space. Combining this noise-denoise cycle with an evolutionary algorithm allows the model to design molecules for specific features.
For example, during the process of designing kinase inhibitors, the model aimed at targeting the active site with high affinity while also trying to reduce off-target effects. This process may be referred to as fine-tuning and reveals further real-life applicability of DiffSBDD-cond in the sense of having focused and effective drug candidates.
Robust Evaluation and Results
To validate their model, the researchers evaluated it on two datasets:
- CrossDocked Dataset: This consists of protein-ligand structures that can be used for learning and testing processes, which are used for the development of systems.
- Binding MOAD: This is a collection of several validated complexes gathered/curated over a period of time through the effort of binding science.
They assessed the quality of generated molecules using metrics such as:
- Vina Score: This represents the affinity towards binding.
- Drug-Likeness (QED): This is calculated for a few molecular features expected to be good/ desirable.
- Synthetic Accessibility (SA): Measures production difficulties by considering economic aspects. This index reflects the price level.
- Diversity: This measures the degree of variety among the generated molecules.
The results were affirmative, and DiffSBDD-cond outbid the existing models Ticke2Mol and ResGen in producing molecules that resemble drugs and have a high binding capacity.
Conclusion
The researchers illustrated how revolutionary AI concepts such as equivariant diffusion models can change the paradigm of structure-based drug design. Set to turn the future of drug discovery upside down, DiffSBDD-cond not only widens this horizon of molecular design but also sets a bar for all future breakthroughs in computational biology.
As the pharmaceutical industry increasingly embraces AI-based technologies, there is hope that approaches such as DiffSBDD-cond could help cut down drug development times and costs and enhance the efficacy of treatments. This is a preview of the future that will be characterized by science and technology coming together to solve some of the most pressing health problems of mankind.
Article Source: Reference Paper | Source codes are publicly available on GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced article. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











-equivariant diffusion to design ligands conditioned on protein pockets for diverse SBDD tasks.){kind=link}